服务咨询热线:
0551-62921227
0551-62921227

新闻中心
 更新时间:2025-08-29
更新时间:2025-08-29 点击次数:518
点击次数:518

第一作者:谢明宇 陈锡燕
通讯作者:杨乐
文章名称:《Iridium single-atoms induced in situ H₂O₂ formation boosting the hydrothermal oxidation of methane to methanol with molecular O₂》
影响因子:6.5
01、老师简介
杨乐老师是中山大学副教授,博士生导师。主持国家自然科学基金、国际合作项目、省自然基金、横向项目等6余项。长期聚焦于生物质高值转化利用、甲烷选择性氧化和二氧化碳加氢制烯烃研究工作。近几年在ACS Energy Letters、Journal of Catalysis、Chemical Engineering Science、Renewable and Sustainable Energy Reviews等期刊上发表论文20余篇,申请国内发明5项。
02、论文研究背景
甲醇是重要的基础化工原料,其工业生产技术涉及通过甲烷合成气的能量密集型两段法工艺,这是一个能源和资本密集型的生产过程。从热力学角度来看,在温和的反应温度下开发甲烷直接部分氧化制甲醇的策略是可取的,因为它更高效、节省资本且环境友好。具体来说,温和的操作模式可以在气田建立,实现就地气体转化为液体,从而便于运输。在过去十年中,甲烷直接部分氧化制含氧化合物已成为一个被广泛研究的课题,其中甲醇是备受关注的目标产物之一。
O₂是甲烷部分氧化制甲醇首先想到的氧化剂。然而,由于O₂是一种高活性氧化剂,这很容易导致过度氧化生成CO₂。为了克服这个问题,可以使用更温和的氧化剂H₂O₂作为替代品。它已被广泛研究用于该反应,以避免过度氧化生成副产物CO₂。然而,H₂O₂价格昂贵且在运输过程中不稳定。理想的情况是通过O₂与H₂反应原位生成H₂O₂,并且已在AuPd@ZSM-5上成功实现,用于高效氧化甲烷制甲醇。然而,同时操作氧化剂和还原剂存在爆炸风险,因此需要极其谨慎小心。关于这个问题,生产 H₂O₂的另一种解决方案是通过H₂O和O₂反应。O₂形成H₂O₂时的吸附构型直接影响CH₄的活化。通常,有三种吸附模型:Pauling型(顶位)、Yeager型(空位)和Griffiths型(桥位)。Pauling型吸附显然有利于H₂O₂的形成,而不会发生O₂中O-O键的断裂,这在Yeager型和Griffiths型模型中很可能发生。因而,基于Pauling型吸附设计催化剂,用于从O₂和H₂O原位生产H₂O₂,用于甲烷部分氧化制甲醇的反应更为合适。
单原子催化剂(SACs)因其高原子利用率而成为成本效益的选择,其应用也已扩展到甲烷的选择性氧化。SACs的一个优势是它们只具有一种类型的活性位点,从而有助于实现高选择性。更重要的是,SACs具有的结构以实现Pauling型吸附,因为每个金属原子都是独立分布的,这使得Yeager型和Griffiths型吸附不可能发生。铱(Ir)基催化剂在甲烷活化方面表现出色。本研究旨在设计基于Pauling型吸附的单原子Ir催化剂(Ir₁/C),用于水热条件下(H₂O为介质)利用O₂实现甲烷高选择性氧化制甲醇。
03、论文亮点/摘要
(1)成功构建了碳负载的单原子铱催化剂(Ir₁/C),其Ir原子与三个碳原子配位形成Ir-C3结构。
(2)理论计算表明,Ir₁/C通过Pauling型吸附促使O₂与H₂O反应原位生成关键中间体H₂O₂(能垒仅0.38 eV),H₂O₂随后解离生成高活性*O物种(能垒0.60 eV),进而高效活化CH₄生成CH₃OH(能垒0.59 eV)。
(3)相反,Ir团簇催化剂(Ircluster/C)通过Yeager/Griffiths型吸附导致O₂直接解离形成强Ir-O键,其CH₄活化能垒较高(1.59 eV)。
(4)Operando红外光谱检测到Ir-OH物种,H₂O₂定量检测(0.1752 mmol g⁻¹)以及顺序进料实验(先O₂后CH₄有效,先CH₄后O₂无效)共同证实了H₂O₂中间体路径。(5)Ir₁/C在温和条件(150 °C, 0.3 MPa CH₄, 0.1 MPa空气, 3 h)下获得17 mmol g⁻¹甲醇产率,显著优于Ircluster/C(6 mmol g⁻¹)。在0.5 MPaCH₄下,甲醇产率高达31.3 mmol g⁻¹,选择性92%,为文献报道值的10倍。
摘要:本研究通过理论指导设计并合成了碳负载单原子铱催化剂(Ir₁/C),实现了水热条件下利用分子O₂高效、高选择性氧化甲烷制甲醇。Ir₁/C的Ir-C₃结构促进O₂进行Pauling型吸附,进而与H₂O反应原位生成H₂O₂中间体(能垒0.38 eV)。H₂O₂在Ir位点解离为*O和H₂O(能垒0.60 eV,速率决定步骤),活化的*O物种高效转化CH₄为CH₃OH(能垒0.59 eV)。实验表征(Operando IR, H₂O₂定量, EPR, XPS, O₂-TPD)和对照实验验证了该反应路径及关键中间体。Ircluster/C则通过O₂直接解离路径进行反应,其CH₄活化能垒(1.59 eV)远高于Ir₁/C。在0.3 MPa CH₄和0.1 MPa空气、150 °C水中反应3小时,Ir₁/C甲醇产率(17 mmol g⁻¹)显著优于Ircluster/C(6 mmol g⁻¹)。优化条件(0.5 MPa CH₄)下,甲醇产率达31.3 mmol g⁻¹,选择性92%。该工作为温和条件下甲烷定向转化提供了新策略。
04、图文解析
1、催化剂精准设计与多维结构表征
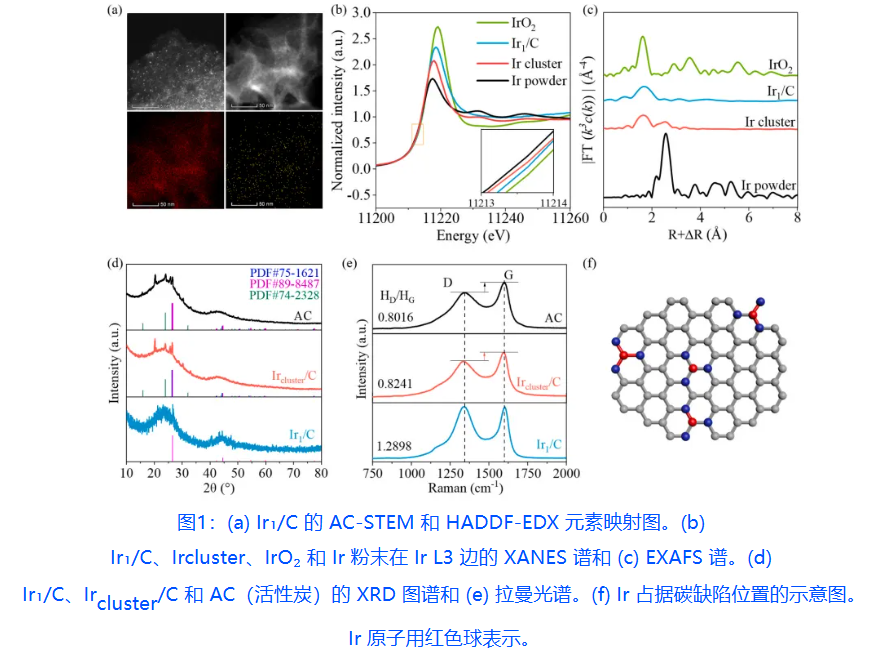
本研究通过高温热解策略(900°C, 10% H₂/N₂)成功构建了碳负载铱单原子催化剂(Ir₁/C)。AC-STEM与元素映射直接证实Ir呈原子级分散(孤立亮点),无团簇聚集,EDX显示Ir信号均匀分布于碳载体。对比催化剂Ircluster/C(500°C热解)则形成4-5 nm大颗粒(TEM统计平均粒径8.56 nm)。采用X射线吸收谱(XAS)表征深度解析电子与配位结构:XANES显示Ir₁/C的白线强度介于Ir⁰与IrO₂之间,表明Ir处于+δ价态(0<δ<4),电子结构经碳载体调变。
EXAFS说明Ir₁/C仅在~2.09 Å处存在主峰(对应Ir-C配位),缺失~2.71 Å的Ir-Ir配位峰,确证Ir以单原子形式与三个碳原子成键(Ir-C₃构型)。而Ircluster/C在~2.70 Å处显现强Ir-Ir配位峰(配位数~2.73),证实团簇形成。
XRD表明Ir₁/C仅在27°出现宽泛衍射峰,表明高温处理破坏碳长程有序性,促进缺陷形成。Raman看出,缺陷指标Iᴅ/Iɢ值从AC的0.80增至Ircluster/C的0.82,并显著跃升至Ir₁/C的1.29,量化证明高温诱导碳缺陷密度大幅提升。Ir₁/C比表面积(~1100 m²/g)低于Ircluster/C(~1500 m²/g),归因于高温导致微孔合并为中孔,但单原子位点的高本征活性抵消此影响。f图为结构模型,清楚观察到Ir单原子锚定于碳层边缘或空位缺陷,与三个碳原子形成稳定的Ir-C₃活性中心,为Pauling型O₂吸附奠定几何基础。
2、理论计算揭示反应路径与能量学本质
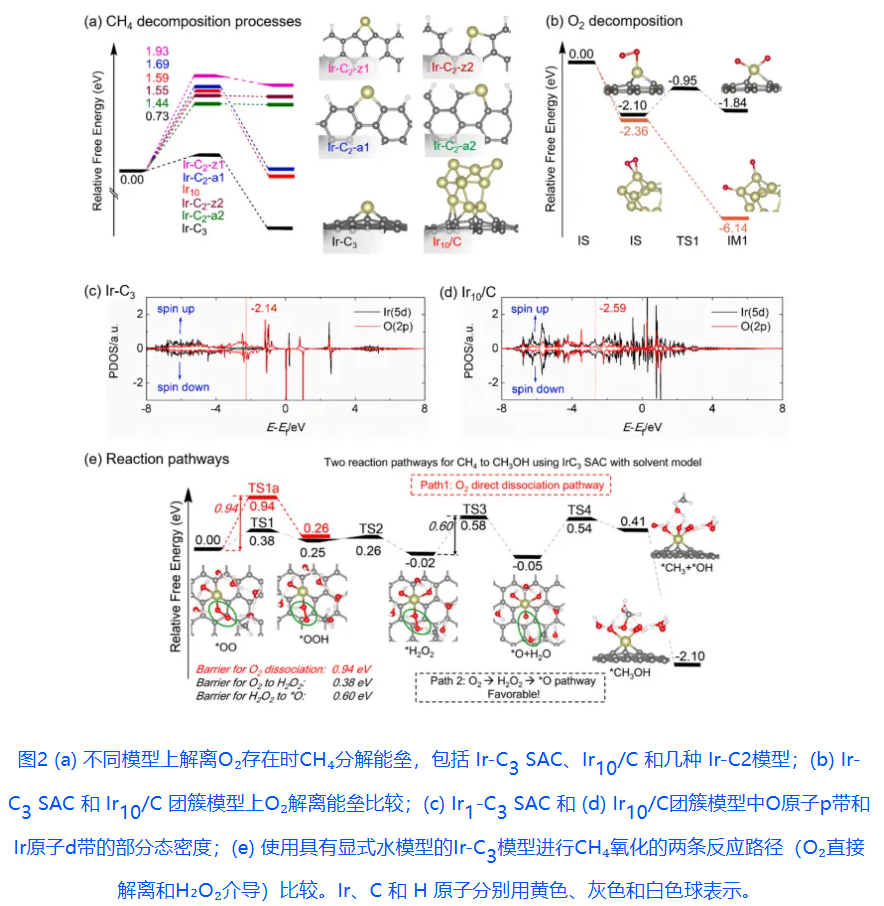
Ir-C3模型对CH₄的C-H键断裂能垒(0.73 eV),源于其强缺电子特性(Bader电荷+0.67 |e|);Ir10团簇模型能垒高达1.59 eV,Ir-C2构型(1.44–1.93 eV)次之,证实Ir-C3为活性中心。
图2b,e揭示了O₂活化路径分化,Ir10团簇中O₂以Yeager/Griffiths型吸附,直接解离无能垒,导致生成强吸附*O物种,引发过度氧化。Ir-C3单原子有两条路径,路径1能垒0.94 eV(显式水模型)。路径2由H₂O₂介导,分为三步:① O₂(Pauling型吸附) + H₂O → *OOH(能垒0.38 eV);② OOH + H₂O → H₂O₂(能垒0.01 eV)→ 原位生成H₂O₂;③ H₂O₂* → *O + H₂O(能垒0.60 eV,速率决定步骤RDS)
总路径能垒(0.60 eV)远小于直接解离路径(0.94 eV)及团簇CH₄活化能垒(1.59 eV),表明H₂O₂路径具绝对动力学优势。PDOS分析显示,Ir-C3上*O物种的p带中心(-2.14 eV)更接近费米能级,显著高于Ir10(-3.50 eV)→ 导致更弱吸附、更强氧化活性,利于CH₄定向转化。显式水模型将H₂O₂形成能垒从1.15 eV(气相)降至0.38 eV,凸显H₂O不仅是介质,更是参与H转移的关键反应物。
3、催化性能与反应中间体的实验验证

Ir1/C具有最高的加氢活性,在150 °C,0.1 MPa条件下获得了17.0 mmol g⁻¹的甲醇产率,Ircluster/C甲醇产率仅为6.0 mmol g⁻¹。优化条件为0.5 MPa CH4后,Ir1/C甲醇产率达31.3 mmol g⁻¹,选择性92%,为文献最高值的10倍。柱状图呈火山型曲线,CH4压力 > 0.5 MPa时产率下降,说明O2分压不足限制*O物种生成。
TPSR-MS显示,先通O2后通CH4,在Ir1/C上同步检测m/z=34(H2O2)与m/z=32(CH3OH)强信号。而先通CH4后通O2则无产物生成,证实反应依赖预活化O2生成*O物种,是H2O2中间体的直接证据。随后采用高灵敏度TMB-HRP酶法,测得Ir1/C液相中H2O2产率0.1752 mmol g⁻¹,为Ircluster/C(0.0584 mmol g⁻¹)的3倍。
150°C下,DMPO捕获的·CH3自由基信号强度:Ir1/C >> Ircluster/C > AC。室温无信号表明C-H断裂为热驱动过程,与DFT能垒(0.59 eV)一致。
在更低O2分压(0.1 MPa空气)下,本研究中Ir1/C的甲醇产率远超文献报道的SACs体系,彰显其原子经济性与氧活化效率。
4. 表面化学与原位光谱揭示动态反应机理
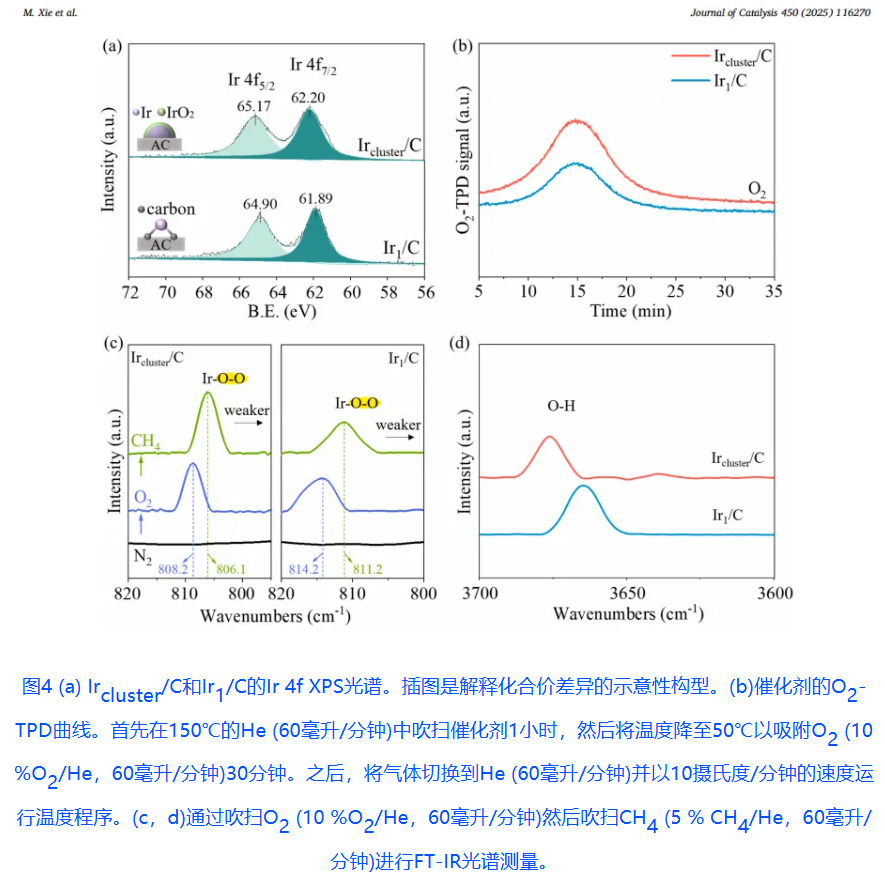
XPS显示Ir1/C(60.8 eV)略低于Ir⁰(61.2 eV),配位诱导电子离域。Ircluster/C(62.1 eV)介于Ir1/C与IrO2(63.5 eV)之间,表明表面Ir原子部分氧化,归因于低配位特性。O2-TPD结果表明Ircluster/C呈现强脱附峰(>400°C)与高吸附量,促进O2解离,而Ir1/C吸附弱且低温脱附,仍维持Pauling型吸附,保护O-O键完整。
FT-IR实时监测O-O 键的动态演变,Ircluster/C的O-O键振动区信号弱且宽泛,说明O2桥位吸附导致O-O键显著弱化。通入CH4后谱带红移~20 cm⁻¹,O-O键进一步拉长,参与C-H活化。~880 cm⁻¹处特征峰归属为Ir-OOH物种(H2O2前体)。Ircluster/C在~3670 cm⁻¹处强峰对应刚性Ir-OH键(与O-O弱化补偿),而Ir1/C此处信号微弱,表明表面主要存在可转移质子的H2O/HOOH物种。
5. 甲烷氧化反应机理探究
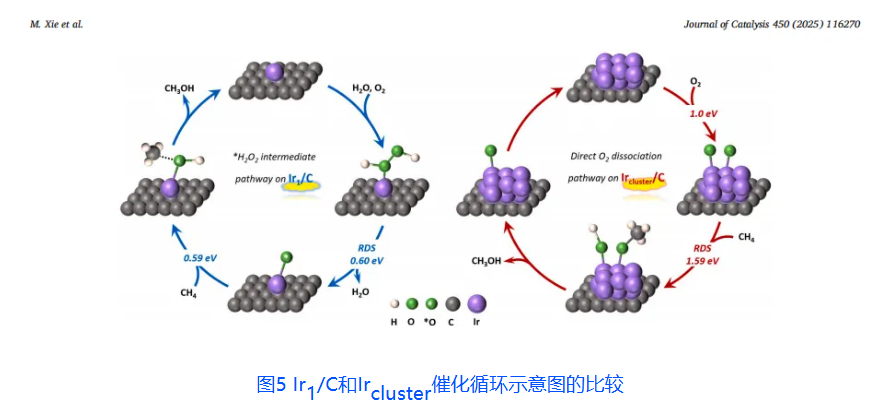
左侧Ir单原子(紫球)锚定于碳缺陷,O2以Pauling型吸附(端基配位),周围水分子参与形成H2O2中间体,低能垒路径生成甲醇。右侧为Ircluster/C,Ir团簇表面O2桥位吸附解离为*O原子(绿球),高能垒路径导致活性与选择性降低。
05本文所用设备
杨乐老师课题组在实验中所用MS系列机械搅拌反应釜由科幂仪器提供,论文中也特别提到安徽科幂仪器有限公司,在此非常感谢老师对科幂仪器的选择和认可。

 当前位置:
当前位置:
 上一个:
上一个: 返回列表
返回列表
